WuXi Biologics
Offering End-to-End Solutions
Research Chemistry
Services
Synthesis of Diverse N-Trifluoromethyl Pyrazoles by Trapping of Transiently-Generated Trifluoromethylhydrazine
Synthesis of Diverse N-Trifluoromethyl Pyrazoles by Trapping of Transiently-Generated Trifluoromethylhydrazine
Bao Li†, Fenglei Xie†, Rui Zhang†, Yaoyi Wang†, Vijaya B. Gondi‡, and Christopher R. H. Hale‡*
†WuXi AppTec Research Chemistry Services, 168 NanHai Road, 10th Avenue, TEDA, Tianjin, 300457, China
‡ Karuna Therapeutics – A Bristol Myers Squibb Company, Chemical Development, 99 High Street, Floor 26, Boston, Massachusetts 02110, United States
*Email: khale@karunatx.com
ABSTRACT
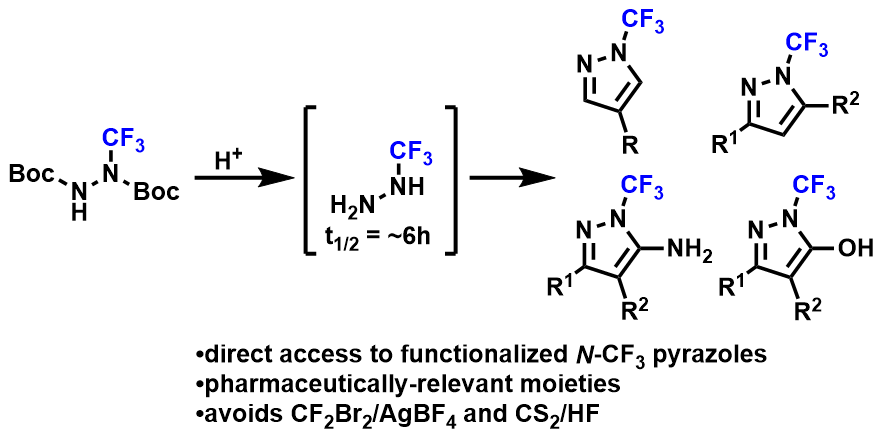
A one-pot synthesis of functionalized N-trifluoromethyl pyrazoles from readily available di-Boc trifluoromethylhydrazine and dialdehydes, diketones, carbonylnitriles, and keto-esters/amides/acids is described. 19F NMR studies were used to characterize the stability of trifluoromethylhydrazine HCl salt in solution and in solid form and identified a short solution-state half-life of ~6 hours. Optimization of cyclization conditions identified DCM, combined with a strong acid, as key to suppress the undesired des-CF3 side-products which formed as a result of the instability of trifluoromethylhydrazine and related intermediates. Despite the short-lived nature of these transient intermediates, their reactivity could be utilized to directly deliver a diverse array of pharmaceutically-relevant N-trifluoromethyl pyrazoles in synthetically useful yields.




How can we help?
Get in touch with an expert.