WuXi Biologics
Offering End-to-End Solutions
Research Chemistry
Services
Chemistry Insights | How to Enhance Success Rate in the Discovery of Targeted Covalent Inhibitors? Design & Synthesis Strategies
Chemistry Insights | How to Enhance Success Rate in the Discovery of Targeted Covalent Inhibitors? Design & Synthesis Strategies
Chemistry is essentially about the creation and disruption of chemical bonds. Although most drugs exert their therapeutic effects through non-covalent interactions with their targets, covalent binding between drugs and their targets has always played a crucial role. For example, widely used drugs like aspirin and penicillin function by creating covalent bonds with their target molecules. In recent years, covalent inhibitor development, particularly for previously considered “undruggable” targets like EGFR, BTK, and KRAS, has seen significant advancement, making this a rapidly evolving area within small molecule drug discovery.
-
- 1. Overview
Covalent inhibitors refer to drugs that form covalent bonds with biological macromolecules such as enzymes or receptors, leading to the inactivation of these targets through covalent bonding. Covalent inhibitors have important advantages over non-covalent counterparts which enable improved potency and prolonged pharmacodynamic effects. These properties enable them to be effective at lower drug doses which improves patient compliance and reduces the likelihood of drug resistance. Additionally, it is critical for covalent compounds to have sufficient selectivity to mitigate the off-target effects. Subsequently, targeted covalent inhibitors were developed and can precisely target specific proteins or enzymes, leading to highly selective therapeutic outcomes.
Over the past decade, advances in structural bioinformatics and medicinal chemistry have significantly enhanced our knowledge regarding the drug development strategies of covalent inhibitors, particularly in oncology. As of December 2023, 12 targeted covalent inhibitors (TCIs) have been approved for cancer treatment [1], targeting six different classes of proteins, including proteasome, Bruton’s tyrosine kinase (BTK), epidermal growth factor receptor (EGFR), exportin 1, fibroblast growth factor receptor (FGFR), and Kirsten rat sarcoma viral protein with a glycine-to-cysteine mutation at position 12 (KRAS G12C). From a structural perspective, it is evident that most of these inhibitors contain the unsaturated carbonyl covalent warhead (Figure 1).
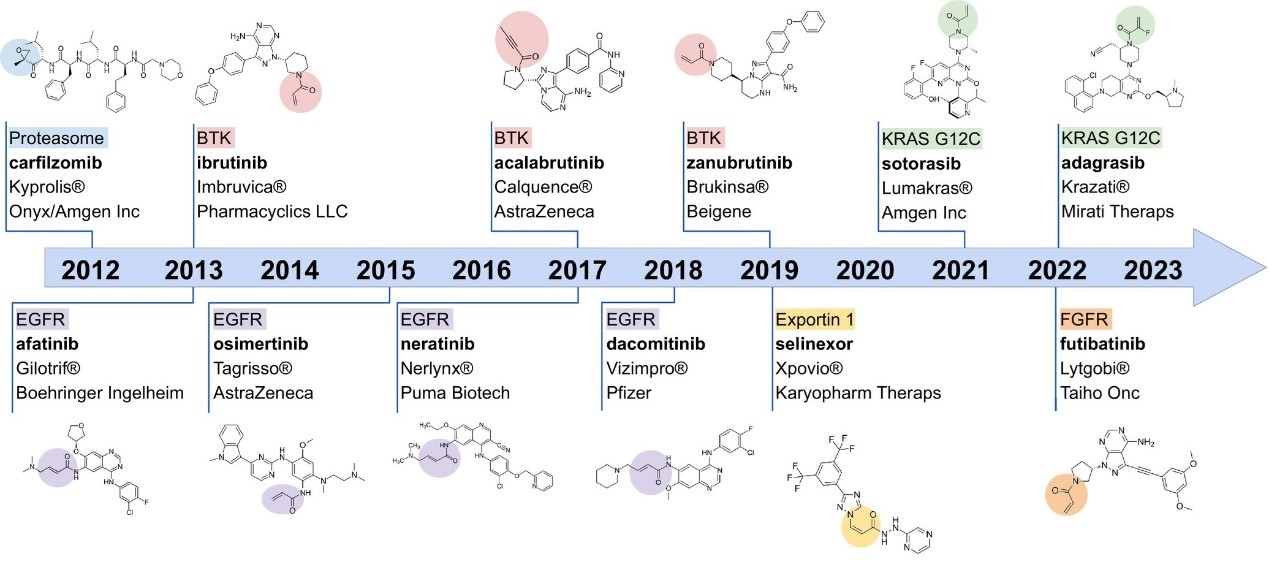
Figure 1. A timeline of targeted covalent inhibitors (TCIs) that have been approved to be marketed by the FDA from 2012 to 2023. [1]
‘
- 2. Discovery Strategies for TCI
The discovery strategies of targeted covalent inhibitors are similar to conventional non-covalent small-molecule inhibitors to some extent. However, there are unique features in the rational design of covalent drugs, such as target selection, identification of appropriate warheads, and target residues. Therefore, the design of general covalent drugs must go through four processes: target identification and selecting the residue; hit identification; binding characterization, and lead optimization.
2.1 Ligand-First Approach
This approach incorporates electrophilic functional groups into known reversible ligands to enhance the inhibition of protein function. The development of the receptor tyrosine kinase EGFR illustrates this strategy.
The reversible, first-generation EGFR inhibitors gefitinib and erlotinib are non-covalent and effective against wild-type EGFR and a limited number of mutant variants. However, the drug-resistance effect is quite prominent in this generation due to their ineffectiveness against EGFR mutations[2](Figure 2).
To overcome this issue, second-generation EGFR covalent inhibitors were developed to effectively inhibit the T790M mutants by introducing an acrylamide reactive group into the quinazoline side chain structure. However, this generation still has higher toxicity side effects due to activity on the wild-type EGFR.
Third-generation EGFR inhibitors refined this approach by incorporating a disubstituted pyrimidine core, selectively targeting the T790M mutant while sparing wild-type EGFR. This selectivity allows higher dosing to improve therapeutic efficacy.
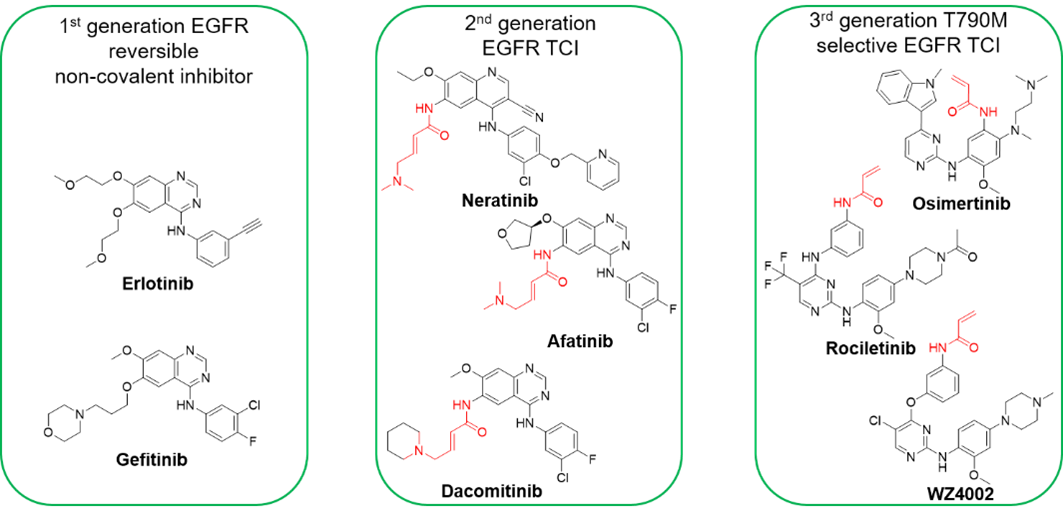
Figure 2. Progression of EGFR inhibitor structures[2]
2.2 Fragment-Based Approach
The establishment and application of the platform of chemoproteomics have revealed thousands of novel interactions between targets and covalent molecules, even for proteins previously considered “undruggable.” This powerful approach allows for high-throughput discovery of pharmacological modulators, enabling the exploration of new protein-protein interactions (PPI, Figure 3). [3]

Figure 3. The roadmap for fragment-based covalent ligand discovery [3]
In the fragment-based discovery strategy, a covalent fragment library is a useful toolbox for discovering both novel protein targets and small-molecule covalent modulators. WuXi AppTec’s Research Chemistry Services (RCS) has been developing a covalent fragment molecule library and has carefully designed and selected a set of covalent fragment molecules from the virtual library. [4]
2.3. Electrophile-First Approach
This method prioritizes the identification of covalent ligands from the outset. A notable example is the discovery of KRASG12C inhibitors. In 2013, Kevan M. Shokat’s team at UCSF discovered compound 6H05 using a disulfide tethering technique, which identified the first ligand for the KRAS protein. The structure of 6H05 was later optimized by replacing the disulfide linker with an acrylamide group as the covalent warhead. This modification revealed that covalent binding of a ligand to KRAS created a hydrophobic pocket on the originally smooth protein surface, allowing small molecules to bind to the protein.
Based on this discovery, researchers systematically optimized compounds from hit to lead, resulting in promising leads like compound 12. Further studies yielded multiple promising preclinical candidates. These groundbreaking results prompted significant investment from pharmaceutical companies, advancing several programs into the clinical stage [3,5] (Figure 4).
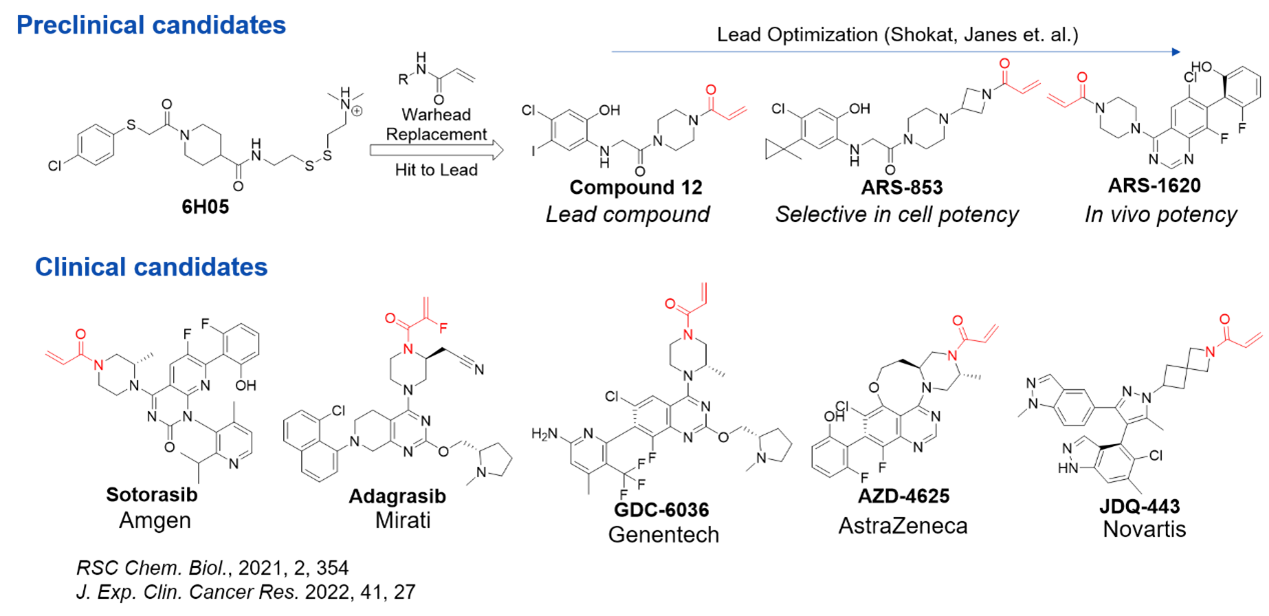
Figure 4. KRAS-targeted covalent drug development[3,5]
3. Covalent Warheads
Covalent warheads are the essential reactive groups in a drug molecule that form covalent bonds with the target protein’s amino acid residues.
The design of covalent warheads is closely tied to specific amino acid residues. In covalent inhibitor development, warheads are categorized by the residues they bind to, with cysteine-targeting warheads being the most common (Figure 5). Cysteine residues are relatively rare, making them highly selective targets for covalent interactions. This classification helps design inhibitors with precise and efficient covalent bonding to specific residues at the binding site.

Figure 5. Various warheads segregated based on interaction with targeted amino acids
According to statistics on marketed covalent drugs, the most common covalent warheads feature the α, β-unsaturated carbonyl, which is widely present in oncology drugs, followed by boronic acid/esters and α-ketoamide present in the antiviral and autoimmune disorder therapeutic area (Figure 6)[6]. In the past three decades, cysteine has been the dominant amino acid residue available for covalent inhibition. As the discovery of TCI evolved, the covalent targeting of tyrosine, lysine, serine, and threonine has attracted more attention in recent years.
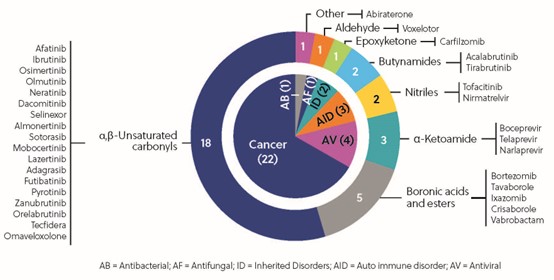
Figure 6. Characteristics of FDA-approved covalent inhibitors [6]
3.1. Unsaturated Michael Acceptors Warheads
All typical acylation conditions are well-used in the covalent modification of parent molecules containing an amino group. Some examples require the aldehyde fragment to form an α, β-unsaturated bond via Knoevenagel condensation (Figure 7).
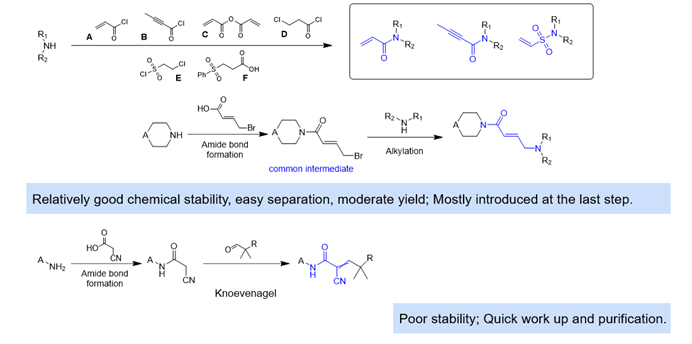
Figure 7. Unsaturated Michael Acceptors Warheads
3.2 Sulfonyl Fluoride Warheads
The sulfonyl fluoride is another classic covalent warhead, considering the broad utility of sulfur (VI) fluoride exchange (SuFEx) in click chemistry. As for the fluorosulfurylation reagent, several easy-to-handle products such as SuFEx-IT and AISF have been commercialized, allowing chemists to pick their preferred method to modify the parent molecule on the OH or amino group. Additionally, the fluoride exchange reaction of sulfonyl chloride provides another robust pathway to access aromatic sulfonyl fluorides (Figure 8).
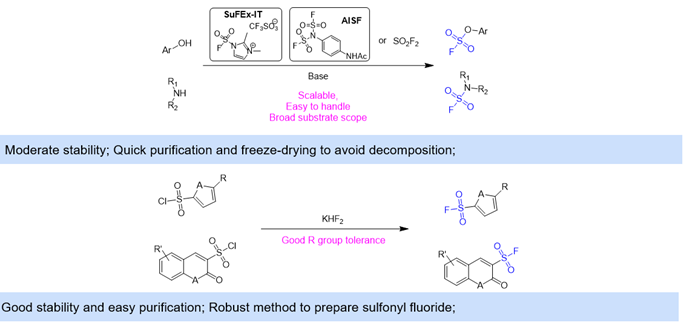
Figure 8. Sulfonyl Fluoride Warheads
3.3 Unsaturated C-X (Carbonyl, Cyano) & Boronic Acid Warheads
The unsaturated C-X bonds such as the carbonyl and cyano group, as well as the boronic acid warhead, can be typically accessed via the reduction/oxidation or dehydration/hydrolysis of corresponding precursors. As these are common functional groups, all the classical synthetic strategies can be adopted and work well on their fitted scope of substrates (Figure 9).
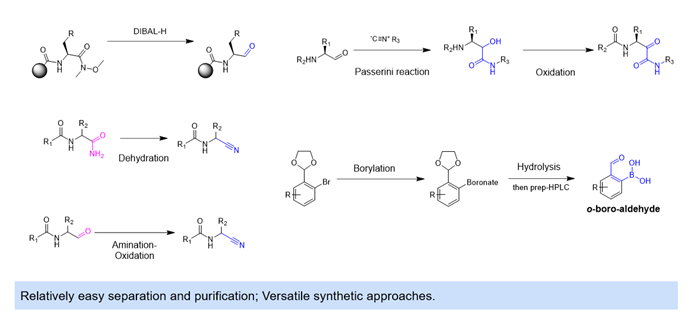
Figure 9. Unsaturated C-X (Carbonyl, Cyano) & Boronic Acid Warheads
3.4 Strained Cyclic Warheads
For the introduction of strained small cycles, it can take several steps of chemistry conversion to synthesize a common intermediate that contains the reactive warhead and to use the intermediate to derivatize the parent molecule (Figure 10). Since the strained small rings are reactive, more caution needs to be paid on the handling of these unstable intermediates.

Figure 10. Strained Cyclic Warheads [7]
Figure 11 summarizes the major types of covalent warheads synthesized via the WuXi RCS covalent chemistry discovery platform. These are all electrophilic reacting groups. The covalent warheads are generally introduced in the later stage or last step in the synthesis of covalent molecules, and their reactivity often causes instability of the compounds.
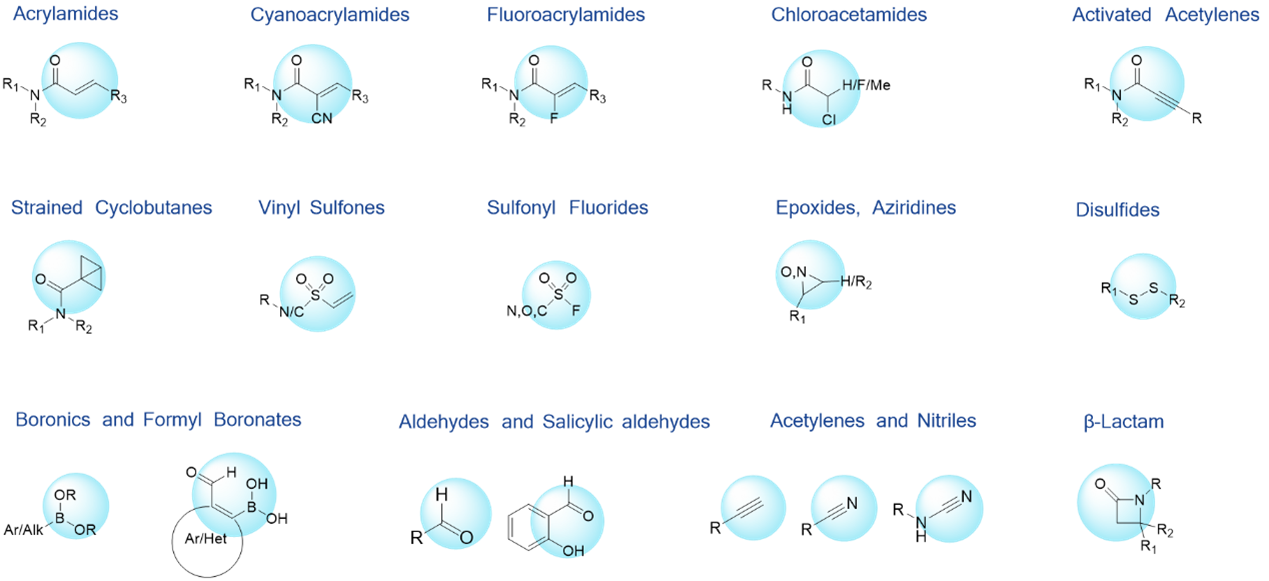
Figure 11. Overview of RCS Synthesized Covalent Warheads
4. Covalent Chemistry Discovery Platform at RCS
Covalent drugs are attracting significant interest in drug discovery due to their unique benefits, potentially leading to more effective therapies and further innovation in various therapeutic areas. Emerging areas of interest include the development of reversible covalent inhibitors, covalent degraders, and covalent targeting of non-cysteine amino acid residues. RCS offers specialized and efficient services to accelerate our partners’ drug discovery process.
RCS has built a comprehensive Covalent Chemistry Discovery Platform offering synthesis, analysis, and purification services for covalent inhibitors (Figure 12). The platform has distinct advantages, including:
- A team of over 1,000 experienced synthetic chemists
- Successful project delivery for over 100 clients or partners
- Expertise in synthesizing more than 40 unique types of warheads, including 17 major types of warheads
- Delivery of over 180,000 covalent compounds since 2018
- Strong expertise in identification of covalent binding using MS-based techniques
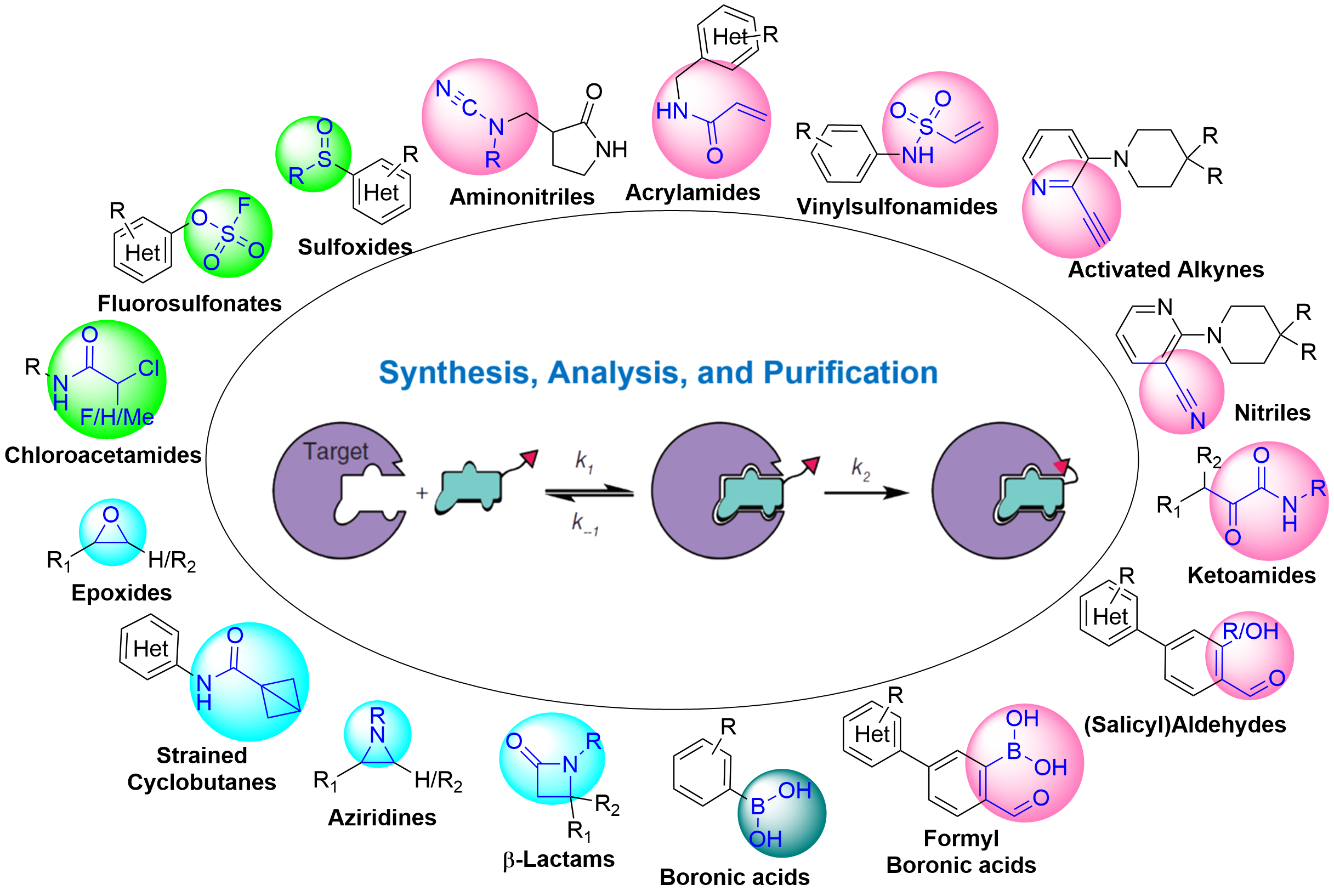
Figure 12. Covalent Chemistry Discovery Platform at RCS
The platform ensures high efficiency in producing diverse covalent compounds while maintaining stringent quality control to ensure optimal stability and purity of covalent drug molecules.
Contact: chemistry_service@wuxiapptec.com
References
[1]) Ngo, H.X.; Wen, Y.W.; Pisupati, S.; Huang, W.; Mandlekar, S. Clin. Pharmacol. Ther. 2024, https://doi.org/10.1002/cpt.3390
[2]) Boike, L.; Henning, N. J.; Nomura, D. K. Nat. Rev. Drug. Discov. 2022, 21, 881-898
[3]) Lu, W. C.; Kostic, M.; Zhang, T. H.; Che, J. W.; Patricelli, M. P.; Jones, L. H.; Chouchani, E. T.; Gray, N. S. RSC Chem. Biol., 2021, 2, 354-367
[4]) Virtual screening identifies hits from extensive chemical space wuxibiology.com/drug-discovery-services/hit-finding-and-screening-services/virtual-screening
[5]) Kwan, A. K.; Piazza, G. A.; Keeton, A. B.; Leite, C. A. J. Exp. Clin. Cancer Res. 2022, 41, art. no. 27
[6]) Covalent inhibitors in strategic therapeutic design | CAS cas.org/resources/cas-insights/rise-covalent-inhibitors-strategic-therapeutic-design#
[7]) Ma, N.; Hu, J.; Zhang, Z.-M.; Liu, W. Y.; Huang, M. H.; Fan, Y. L.; Yin, X. F.; Wang, J. G.; Ding, K.; Ye, W. C.; Li, Z. Q. J. Am. Chem. Soc. 2020, 142, 6051-6059
Disclaimer: This presentation is solely for discussion and informational purposes. It does not constitute an offer to provide the compounds/technologies mentioned. Any order placed will be subject to a thorough IP risk assessment. We will only accept orders for synthesis services if it is determined that no third-party intellectual property rights are infringed.




How can we help?
Get in touch with an expert.